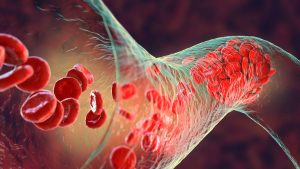
Over 20,000 individual IVC filter lawsuits have been filed since 2012. Many claims have been settled but strong suits are still pending. Our firm continues
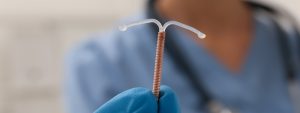
Traducido por Jose De Jesus Sanchez Los dispositivos intrauterinos (DIU) son un método anticonceptivo comúnmente utilizado por las mujeres en todo el mundo. Sin embargo,
An IVC (Inferior Vena Cava) filter is a small, cone-shaped medical device that is commonly implanted into a patient’s inferior vena cava, which is the

By Mia Wang What is a Port-a-Cath? A port-a-cath is used to give fluids, blood transfusions, chemotherapy, antibiotics, food, and other drugs through the blood
The Paragard Multidistrict Litigation is moving forward after US District Court Judge issued the latest scheduling order concerning deadlines for discovery, including disclosure of experts

By Benjamin Pace In a significant development, two members of the Senate Judiciary Committee, Senators Dick Durbin (D-Ill.) and Richard Blumenthal (D-Conn.), are urging a
Megadyne has recently received reports of 63 injuries (and counting) related to their MEGA 2000 and MEGA SOFT Reusable Patient Electrode devices.[1] The list of
The Actions against Bard seek to hold the device manufacturer liable for injuries caused by their wrongful conduct in connection with the design, development, manufacture,
Also commonly referred to as Injection Parts or Port-a-Catheters, the Bard PowerPort products at issue in these lawsuits are implantable vascular access devices designed to
Paragard, a type of intrauterine device (IUD) manufactured by Teva Pharmaceuticals, has been the subject of numerous lawsuits in recent years. These lawsuits have claimed that the
Last week the annual shareholder meeting of Johnson & Johnson took place at its headquarters in New Brunswick, New Jersey. Despite the proclamation of approvals
By Maria Markou, Esq. Honorable Judge Brian Martinotti supervises in a multi-county litigation (MCL) in Bergen County, where more than half of the nearly 1,500
By Paul D. Rheingold A respected securities analyst, Northland Securities, Inc. released a March 2013 advisory regarding Intuitive Surgical’s stock valuation. Due to the shortcomings
Many patients and physicians in New York are taking note of claims of injuries caused by robotic medical devices that are not being reported to
A class I recall was issued in April, 2015, for all HeartWare Ventricular Assist Systems (VAS) currently in use. The HeartWare VAS helps deliver blood
By: Kelda Doherty It is no surprise to our firm that Johnson & Johnson, the largest manufacturer of health care products in the world, has
A recent Wall Street Journal report found that 14 deaths were tied to Medtronic’s SynchroMed drug-infusion pumps. The Department of Justice (DOJ), in conjunction with
FDA announced a Class I Recall of the Tiger Paw System II, produced by Maquet Medical Systems. The Class I is the most serious FDA
The FDA has issued its most serious recall, Class 1, on MicroPort Orthopedics’ Profemur Long Cobalt Chrome 8 Degree Varus/Valgus Modular Neck devices. The Shanghai-based
by Maria Markou Our firm specializes in representing people with serious injuries as a result of harmful medical devices and drugs. Recent FDA recall statistics
by Maria Markou The Rheingold product liability trial litigation team has represented many injured clients and wrongful death cases against Johnson & Johnson: DePuy Pinnacle
By: Indhira Benitez As recently reported, Minnesota anesthesiologist, Dr. Scott Augustine, has been campaigning to remove the device he helped create off of the market.
By: Kelda Doherty The United States Judicial Panel on Multidistrict Litigation (JPML) has this month ordered that pre-trial proceedings concerning the Stryker’s Rejuvenate and ABG
Heater-cooler devices are used during cardiothoracic surgeries, as well as other medical and surgical procedures to warm or cool a patient to improve health outcomes.
Ever since the first approval of medical devices in 1976, the FDA has been struggling to keep up with the pace of technological advancement. Many
Every day we hear about dangerous drugs and medical devices. The U.S. Food and Drug Administration (FDA) routinely reports on investigations, warnings, injuries and recalls.
A lawsuit is underway in California Superior Court against DePuy, Inc., a subsidiary of Johnson & Johnson for their defective ASR hip implant system. Over
How do prescription drugs get recalled? How do problematic artificial hips get fixed? Many manufacturers imprint a seriel number on their devices – the problem
Retrievable IVC Filter Liability Attorneys Retrievable IVC filters are designed to catch blood clots in patients before they reach the lungs. The IVC or Inferior
Johnson & Johnson may face a sequel to its 2010 $3 billion recall of ASR all-metal artificial hips. The company’s DePuy Orthopedics unit and its
The system used by the Food and Drug Administration (FDA) to grant market approval of medical devices is slated for some updates in 2011. The bureaucracy’s
The FDA recently released an alert to healthcare professional regarding the Lariat Suture Delivery Device, manufactured by SentreHEART. The device uses a tiny lasso like
Pharmaceutical giant, Boston Scientific Corporation, recalled its global supply Chariot Guiding Sheaths which total 7,000. The recall was initiated because parts of the device can